
Dr Alan Richardson
- Title
- Reader in Pharmacology
- Location
- Hornbeam 1.08
- Role
- Pharmacology Lead and Group Lead of Cancer research group
- Contact me
- Via email
- Phone
- +44 (0)1782 733571 / +44 (0)1728 674424
- a.richardson1@keele.ac.uk
Biography
I joined Keele in 2008. Following a PhD and post-doctoral work in pharmacology at Cambridge, I completed my post-doctoral training at the University of Virginia, USA. Following several years leading drug discovery programs in industry (Johnson & Johnson, Belgium and OSI pharmaceuticals, Oxford) I returned to academic research at the Institute of Cancer Research in London. At Keele, I lead teaching of pharmacology including option topics focusing on oncology molecular therapeutics. I also make teaching contributions to the School of Life Sciences and the School of Medicine. I am currently third year tutor. My research interests focus on discovering new therapeutic targets and drugs for the treatment of ovarian cancer. I have received funding from charities and research councils.
Research and scholarship
Research
Research group: Cancer (Group Lead)
Research interests
The goal of research in my group is to help discover new treatments for cancer. During my career I have been fortunate to make significant contributions to several projects that have resulted in drugs entering clinical trials. I (or the group I led) were the first to identify a biological function for focal adhesion kinase, clone full length Akt3 and discover some of the first Akt inhibitors, demonstrate that a class of drugs called BH3 mimetics sensitize ovarian cancer to chemotherapy, show pitavastatin is likely to be the most successful statin for cancer therapy and during the pandemic to show that fenofibrate inhibits SARS-COV2 infection in cell culture models.
I currently trying to discover new therapies for breast and ovarian cancer. The fundamental research performed into cancer biology over the last 30 years or so has been translated into new therapies that are now becoming available to treat patients. These “targeted therapies” differ from most traditional chemotherapy by exploiting specific defects in cancer cells. Cancer cells may become overly dependent on one particular signalling pathway and so are susceptible to drugs that target those pathways. Non-cancerous cells which are less dependent on these pathways are consequently less sensitive to the effects of the targeted therapies. As a consequence, targeted therapies often have milder side effects than traditional chemotherapy.
In some cases, targeted therapies have been very successful, but in other cases the drugs have not been as successful as originally hoped. There are several reasons for this. One of the key reasons is that tumours are heterogeneous – the cells in tumours are not all the same and different cancer cells may be driven by different defects. Consequently, one drug targeting a single defect may not be able to kill all the cancer cells in the tumour. A second reason for some drugs underperforming is that the cancer cells can “evolve”. Cancer cells can mutate and become resistant to a particular targeted therapeutic drug. This gives the cells with the mutation a selective advantage because cells with the mutation continue to grow in the presence of the drug while cells without the drug are killed. A third reason is that tumours may be caused by defects in several pathways, so a targeted therapeutic drug that inhibits a single pathway may not be sufficient to kill the cancer cells.
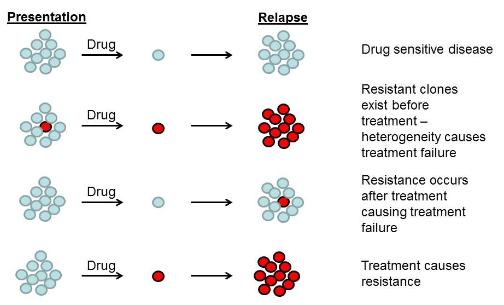
The diagram shows the why cancer therapies may fail. Cancer cells (blue circles) maybe sensitive to therapeutic drugs, but other (red circles) may be resistant.
There are several potential solutions to these problems. One is to use combinations of drugs which inhibit several pathways at the same time. Another solution is to target essential cellular processes that are used by a large proportion of the cancer cells. Lastly, it may be possible to find drugs which make traditional chemotherapy more effective. Research in my group is making use of all these strategies.
Identifying and targeting the pathways involved in resistance to chemotherapy
One of the barriers to the successful treatment of cancer patients with chemotherapy is that the tumours can evolve to become resistant to the drugs. We have previously conducted a RNAi screen to identify genes which confer resistance to carboplatin and paclitaxel (Vidot et al), drugs commonly used to treat ovarian cancer. One of the genes we identified in the screen was autotaxin. We have gone on to develop novel delivery strategies for drugs that inhibit autotaxin (Fisher et al).

The diagram shows the effect of knockdown of 90 different genes, known to be over-expressed in ovarian cancer, on the sensitivity of 6 different cell lines to carboplatin (“C”) and paclitaxel (T). Data from Vidot et al.
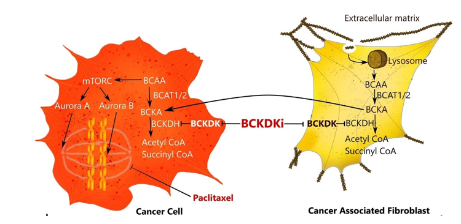
Another gene to emerge from the screen was “Branched-chain keto-acid dehydrogenase.” This kinase regulates the metabolism of branched-chain amino acids (BCAA) and we found that its inhibition with several different drugs or siRNA sensitized both breast and ovarian cancer cells to paclitaxel chemotherapy. We also identified the mechanism underlying this – inhibition of BCKDK promotes BCAA metabolism, leading to the inactivation of t he mTORC1 and Aurora pathways that regulate M phase cell cycle checkpoints activated by paclitaxel (see diagram). We have subsequently identified novel BCKDK inhibitors (Ibrahim et al). Several other exciting hits from the screen remain to be explored.
Statins
Statins inhibit HMGCR, an enzyme involved in the biosynthesis of cholesterol and isoprenoids. There are numerous reports that statins can kill cancer cells in the laboratory, and studies looking at patients taking statins to control their cholesterol also hint at a reduced risk of cancer. However, when statins have been tested in prospective clinical trials in cancer, they have so far largely been unimpressive. We have found three reasons that are likely to explain why these trials have failed in the past and, importantly, how clinical trials should be designed in the future to overcome these problems.
- Pitavastatin is a unique statin which is more likely than other statins to be successful as a cancer treatment. It is the only statin which is both hydrophobic (which makes the drug potent) and has a sufficiently long half-life to allow continual inhibition of HMGCR with a reasonable interval between doses.
- A suitable dosing regimen needs to be used. Relatively high doses of pitavastatin are likely to be necessary. The dose of statins used to treat elevated cholesterol does not provide a sufficiently high drug concentration to kill cancer cells. The interval between doses needs to maintain continual inhibition of HMGCR (Robinson et al) and in the case of pitavastatin this can be achieved with twice daily dosing. This regimen brings with it a risk of causing myopathy which needs to be monitored.
- Diet needs to be controlled. Statins kill cancer cells by preventing the production of geranylgeraniol but this is found in some foods, bypassing the effect of the statins. “Ensure” is one potential food replacement that lacks geranylgeraniol and which patients could use while receiving pitavastatin therapy.
When all the factors are taken into account, we have shown that we can cause regression of tumours in mice (deWolf et al).
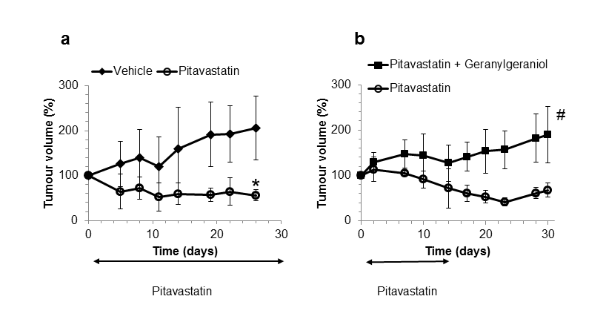
The diagram (left) shows regression of Ovcar-4 tumours in mice treated with pitavastatin and on a diet of “Ensure”. The second diagram (right) shows that if the diet contains geranylgeraniol, pitavastatin no longer shrinks the tumour. From deWolf et al.
We have also shown that zoledronate, a drug normally used to treat bone disorders, potentiates the activity of pitavastatin, and this may reduce the dose of pitavastatin that is necessary to treat cancer (Abdullah et al).
Autophagy
Autophagy is the process by which cells can recycle damaged cellular components. Cancer cells use autophagy to survive nutrient deprivation, hypoxia and low pH and they can also become dependent on autophagy for survival under oncogenic stress. Several studies have also demonstrated that pharmacological or genetic inhibition of autophagy sensitises cells to anti-cancer drugs. We are focusing on a key step in autophagy, the formation of a complex between Atg16 and Atg5 (shown in the figure). We have developed novel autophagy assays and this has allowed us to identify compounds which inhibit the interaction of Atg5-Atg16 interaction. We are currently optimizing these compounds with the goal that these will subsequently be tested in patients with cancer.
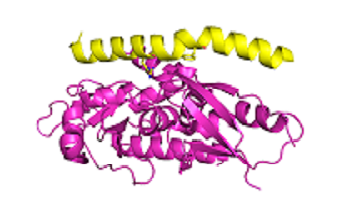
The diagram shows the interaction of Atg5 (magenta) and Atg 16 (yellow) Data from Kim et al.
Creating new research models
It sounds shocking, but 90% of drugs that enter clinical trials are not successful. Part of the reason must be that the way we test drugs in the lab is not predictive of what happens in patients. My current goal is to develop new methods to change the paradigm by which new drugs are discovered and improve this situation. This will lead to better and cheaper drugs.
Public outreach
I have created the “Why do scientists do what scientists do” website to explain to a lay audience some of the things scientists get up to. It is also a useful introduction for students of science. I have also given several public talks explaining the issues in treating cancer (eg at Keele's 75th anniversary).
Funding
Funding for work in our group has been gratefully received from:
- Medical Research Council
- Wellcome Trust
- The Higher Council for Education Development in Iraq
- Ministry of Higher Education and Scientific Research (Iraq)
- Keele University
- The North Staffordshire Medical Institute.
We welcome offers of funding to support our research – nationally, only 10-20% of grant applications are currently funded. If you would like to contribute to our research to discover new cancer treatments, please get in contact. Donations may be tax deductible. Thank you!
Education
I have also generated several innovative teaching methods.
- Together with Luke Bracegirdle, I have developed "KUiz" a tool to allow academic staff to create rapidly a quiz for students that can be viewed on a smartphone or PC.
- I use the Keele KAVE to teach molecular pharmacology in a 3D virtual environment, and I have shown that this improves student learning
- I have led the development of a synoptic assessment that allows students to integrate and entire year's learning
- I have developed an inexpensive method for simulating drug-receptor interactions in a classroom
Publications
Head of School:
Professor Heidi Fuller
Email: h.r.fuller@keele.ac.uk
School address:
School of Allied Health Professions and Pharmacy
MacKay Building
Hornbeam Building
Keele University
Staffordshire
ST5 5BG
Enquiries:
Placements team: sahp.practiceplacements@keele.ac.uk
Postgraduate course admin team: sahp.postgraduate-admin@keele.ac.uk
Undergraduate course admin team: sahp.admin@keele.ac.uk
Undergraduate enquiries:
Email: enquiries@keele.ac.uk
Tel: +44 (0)1782 734010
Postgraduate enquiries:
Email: sahp.postgraduate-admin@keele.ac.uk
Keele Centre for Medicines Optimisation (KCMO)
Tel: +44 (0)1782 733831 / 734131
The Virtual Patient project enquiries:
Contact our Digital Development team:
Email: pharmacy.digital@keele.ac.uk
Pharmacy postgraduate enquiries:
Please contact the CPD4ALL team: cpd4all@keele.ac.uk
Pharmacy email: phab.postgraduate@keele.ac.uk

